Philosophy
“.. so much progress [in biology] depends on the interplay of techniques, discoveries and new ideas, probably in that order…” – Sydney Brenner
Technology drives biological discovery. While we could make significant progress in deciphering biology using existing approaches, leaps in progress come with innovations that enable other researchers to look at their systems in new ways. By creating the experimental and computational approaches of tomorrow and enabling other scientists to adopt and extend our work, we will maximize our impact.
“The best way to have a good idea is to have lots of ideas” – Linus Pauling
Innovation starts with creativity. It is important to keep our goal in mind as we brainstorm creative solutions that solve the problems of today so we can reach it. Not every idea will work or even be worth pursuing, but by having numerous threads leading to our goal, we are poised to take advantage of opportunities as they arise.
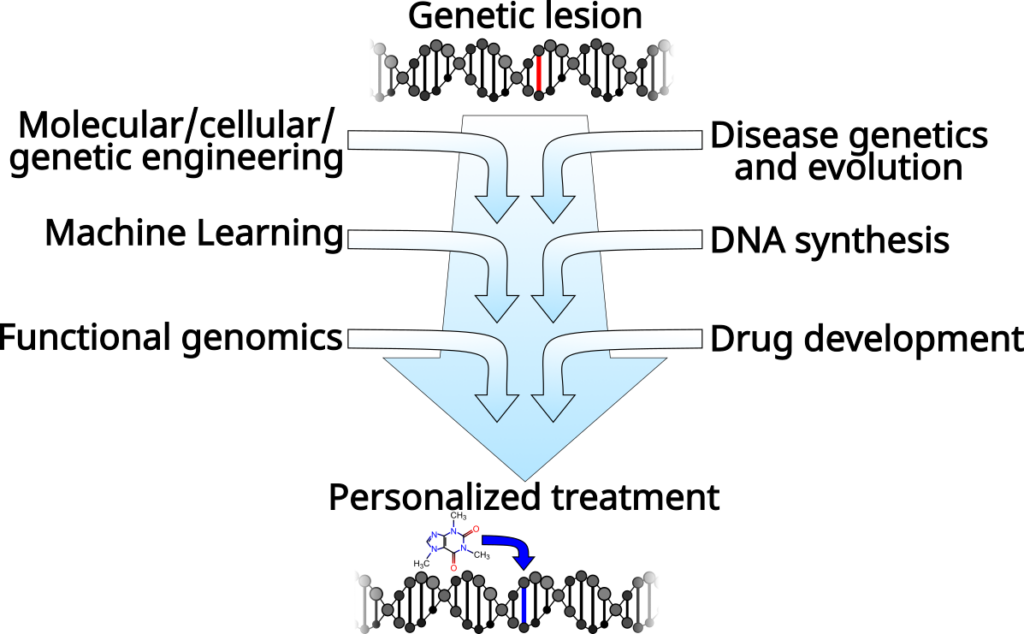
From fundamental biology to therapeutics
Our aim is to create a pipeline to go from genome sequence to personalized therapy. In particular, we are focused on the regulation of gene expression, which is fundamental to our efforts to understand and engineer biological systems, and is a critical aspect of nearly every disease. Our research includes DNA synthesis and omics technology development, algorithm and computer model design, and drug design and discovery. However, this all lies along a clear path from understanding how genomes are regulated so that we can treat diseases that originate in altered gene regulation.
A major part of our current efforts aims to understand how gene sequence encodes for gene regulatory programs. Each gene has regulatory regions that control when and where that gene is expressed. These regulatory regions, including promoters and enhancers, are interpreted by proteins called Transcription Factors (TFs). TFs physically interact with the DNA in a sequence-specific way and work together to produce a regulatory output. Regulation not only includes when, where, and how much a gene should be transcribed into mRNA, but also what regions of the genome should be transcribed.
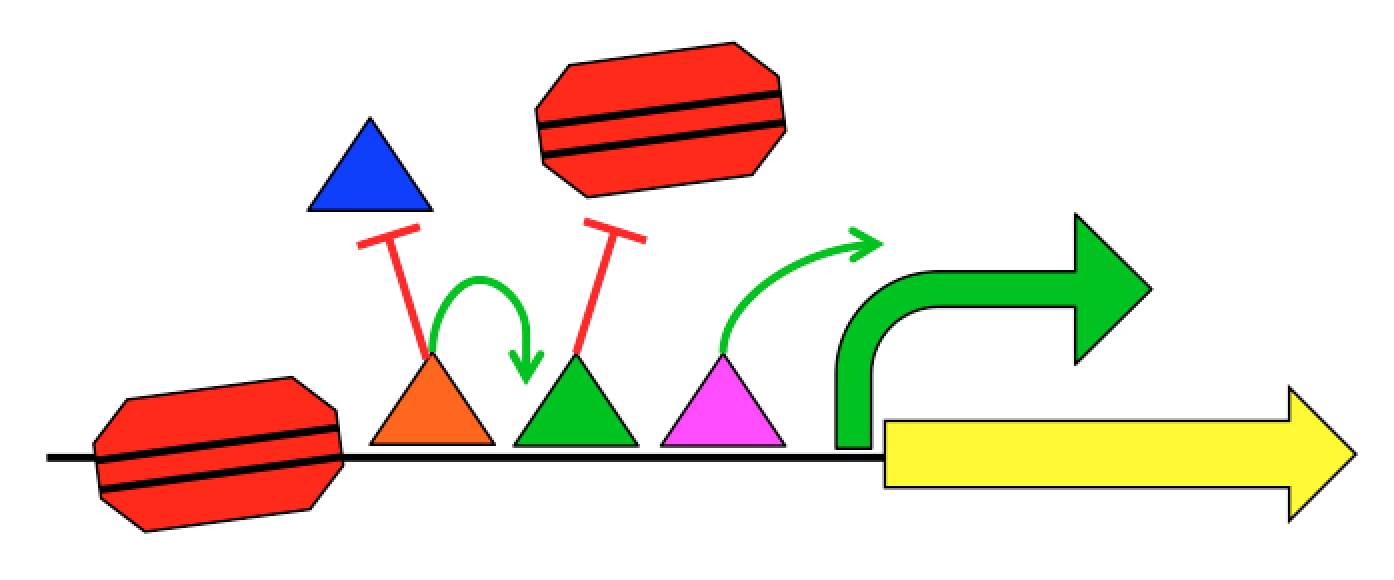
We know roughly how TFs regulate expression, and it can be exceedingly complex. TFs can interact with the transcriptional machinery to regulate transcription initiation, but they can also physically interact with each other and with the nucleosomes that package the genome. This complexity, combined with the limited training examples provided by nature, has limited our ability to create predictive gene regulation models. For instance, the 1639 human TFs can potentially interact in hundreds of millions of different ways, but the human genome is only 3 billion bases long with about 100,000 regulatory regions per cell type. Our group aims to develop the quantitative models necessary to understand gene regulation through synthetic genomics and machine learning. We create new examples of regulatory sequences at genome scale, and apply advanced machine learning approaches to learn how they work.
Why should we care how gene sequence encodes for expression? Gene regulation is fundamental to most of biology, from cellular processes to organismal development. We are particularly interested in two applications: (1) regulatory element design and (2) regulatory genetic variation.
A model that predicts gene regulation from sequence could be used to design sequences with specific regulatory properties. For instance, we could engineer a synthetic enhancer that would turn on a “self-destruct” gene when our cell comes into contact with a cancer cell. Many cellular therapeutics could benefit from precise control of gene expression.
Complex inherited diseases, like autoimmunity and heart disease, are caused, not by one mutation, but by a constellation of common genetic variants across the entire genome, with each variant contributing only a little to disease risk. The vast majority of risk variants lie in the regulatory regions of the genome. If we had models that understood how sequence encodes for expression, this would shed much needed light on how these variants contribute to disease. It is important to know both which genes a variant affects and in what cell type and condition, but also what TFs have had their binding altered by this variant. This could allow us to design personalized therapeutics to correct the misregulation.
Big data for regulatory genomics
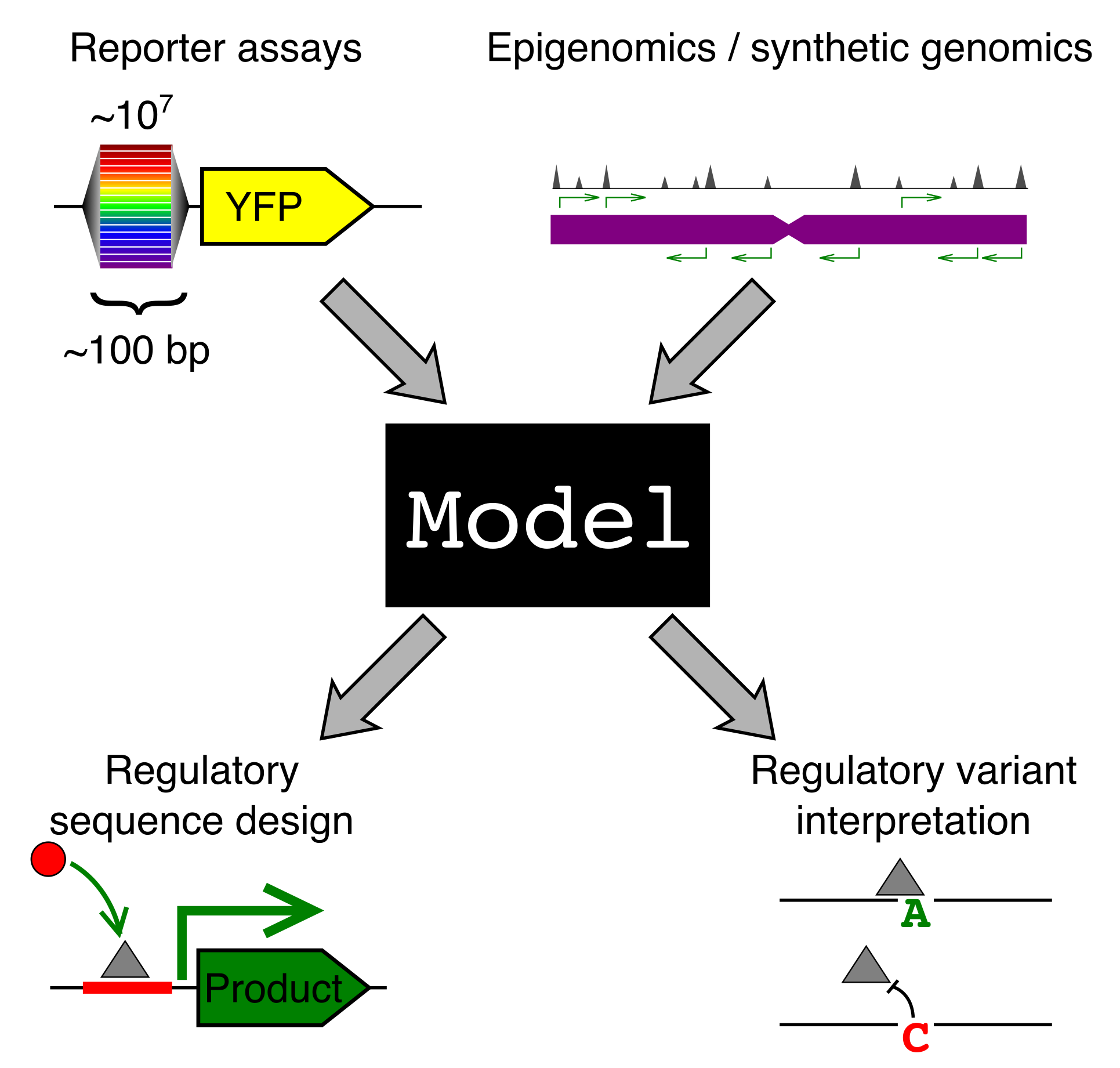
A computational gene expression model can only be created with training data. For reasons that are explained fully in our 2019 paper, random DNA (bases randomly selected from the four possibilities) turns out to be a great way to generate training data at massive scale.
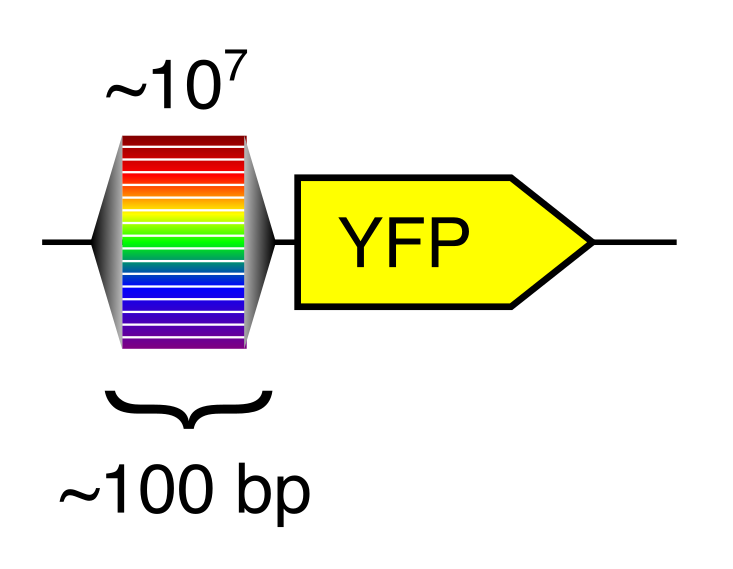
Random DNA encodes for diverse expression levels because of the diverse TF binding sites that occur there by chance. Each random sequence encodes for a specific and reproducible expression level.
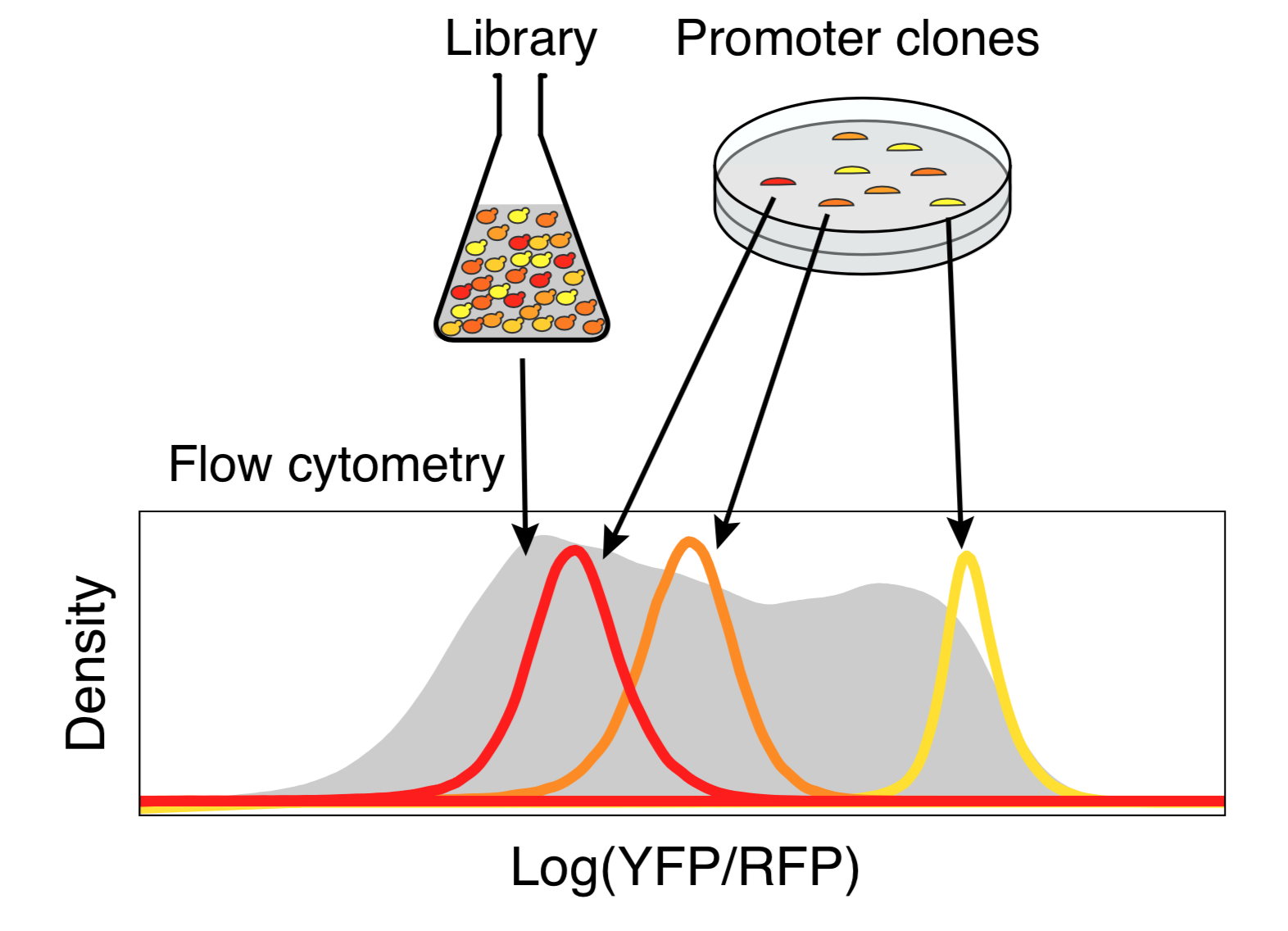
Any two random oligonucleotides have a negligible chance of being similar, and so the chances of training/test violations is minuscule (this is a significant problem with natural sequences). Random DNA is also easy to synthesize and clone at massive scale. Together, this makes random DNA ideal as a source of training data. In our proof-of-concept, using yeast as a model, we were able to measure the regulatory activity of a human genome’s worth of new regulatory DNA in each experiment.
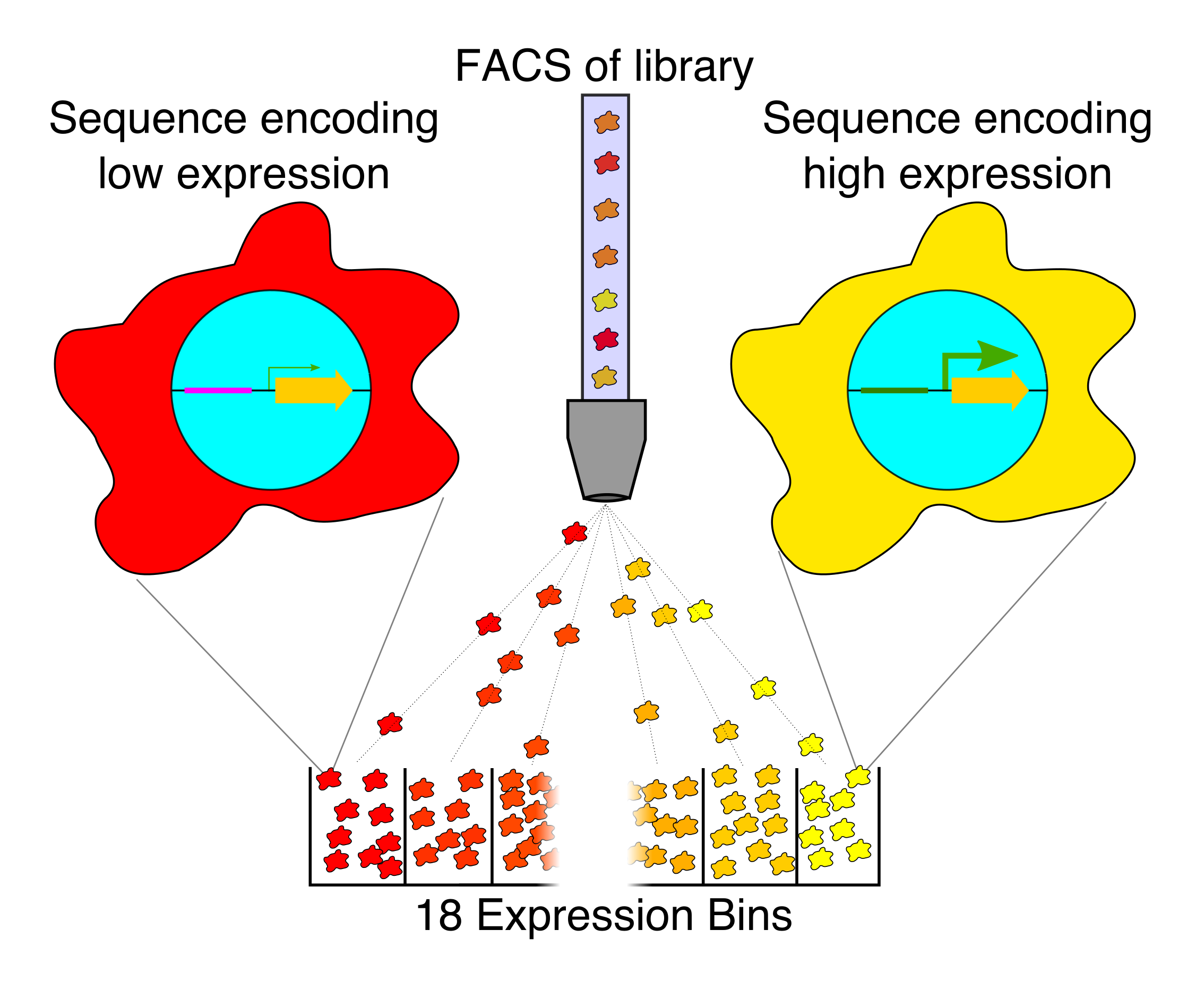
Our current research in this area is focused on exploring different ways of generating ideal machine learning datasets for training gene regulatory models in human cells. For instance, random DNA, partially-random DNA, randomly mutated DNA, making bigger random elements, expanding the scale of our regulatory assays, and creating new types of regulatory assays.
Modeling gene regulation
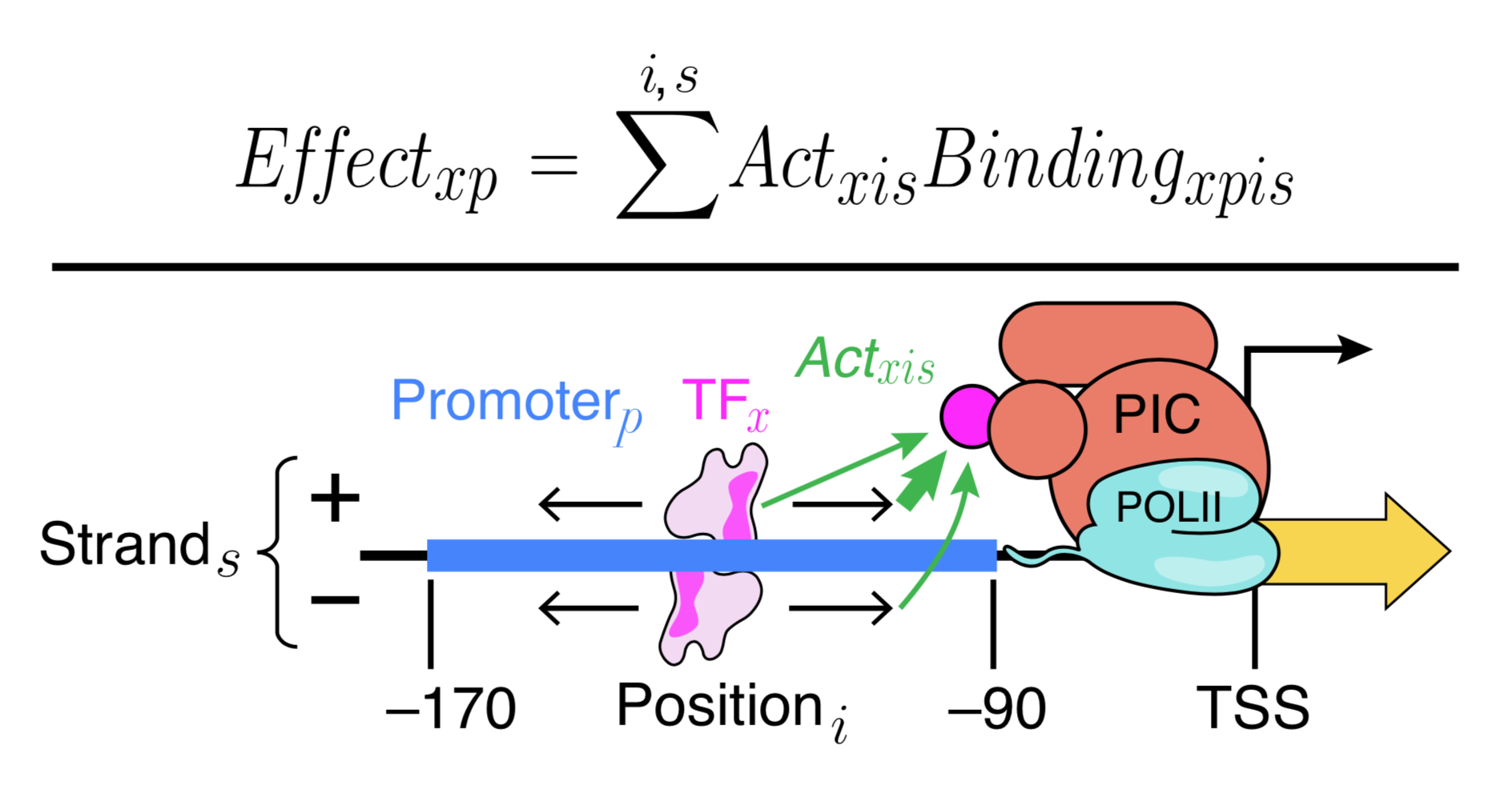
Generally, our models fall into two types that serve different purposes. We use Deep Convolutional Neural Networks to learn highly expressive models that can capture arbitrary biophysical phenomena and have excellent predictive power. Because neural networks are very difficult to interpret, we also make interpretable models (e.g. Physics-informed neural networks) that aim to capture known biophysical phenomena in a quantitative model. We learned a lot of new biochemistry from our interpretable yeast models, but we also know that there is much more to learn. Our interpretable models could not explain expression as well as Deep Convolutional Neural Networks. By comparing the two models, we can identify where our interpretable models fall short, and add additional parameters to include the new biochemistry. This will allow us to learn new regulatory mechanisms, and build ever more accurate models. We are also continuing to develop even better neural networks, including with the community through a recent DREAM Challenge.
The evolution of gene regulation
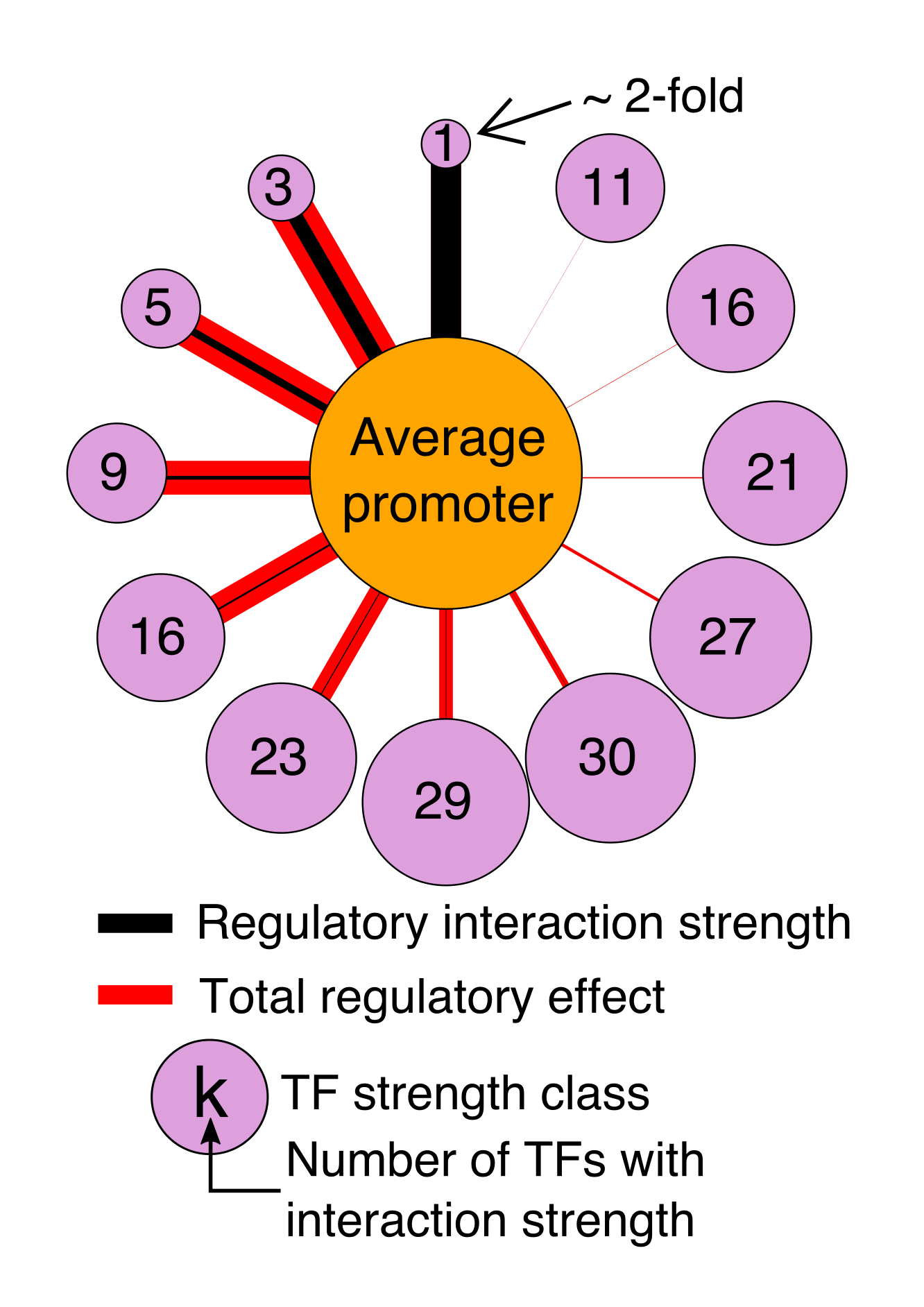
Regulatory DNA sequences evolve very rapidly compared with protein sequence. Surprisingly, gene regulatory programs can be conserved in spite of widespread divergence of the regulatory DNA. The regulators themselves, TFs, also diverge very slowly. In fact, same regulatory element can often encode the same expression program in mice and zebrafish, indicating that the TFs are interpreting the DNA the same way in both species. Using computational models, we can explore the evolution of gene regulation.
How do regulatory sequences change under different types of selection? How do new TFs evolve? How do gene regulatory networks change over time? We aim to answer these questions and more.
Gene regulation in disease
An important application of our work is to understand how genetics and epigenetic factors affect gene regulation, and contribute to disease risk and severity. For example, mapping open chromatin can identify genomic regions that are differentially active between disease and healthy, leading to potential disease markers, and treatments. Profiling how cell states and types change with aging can highlight cells important in age-related disease. Finally, we can seek to understand the role non-coding genetic polymorphisms play in disease by profiling their regulatory activity in relevant cellular models.
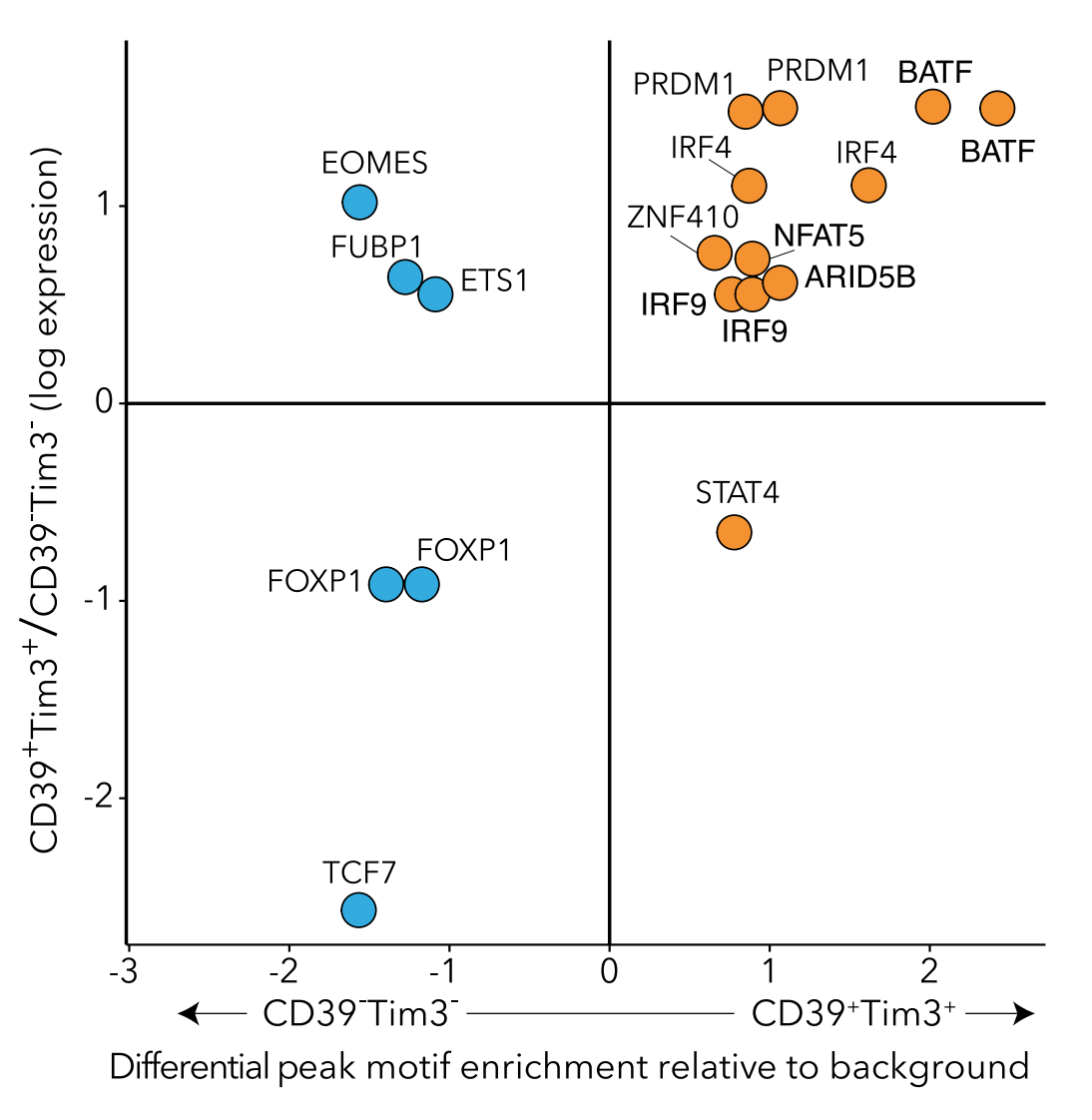
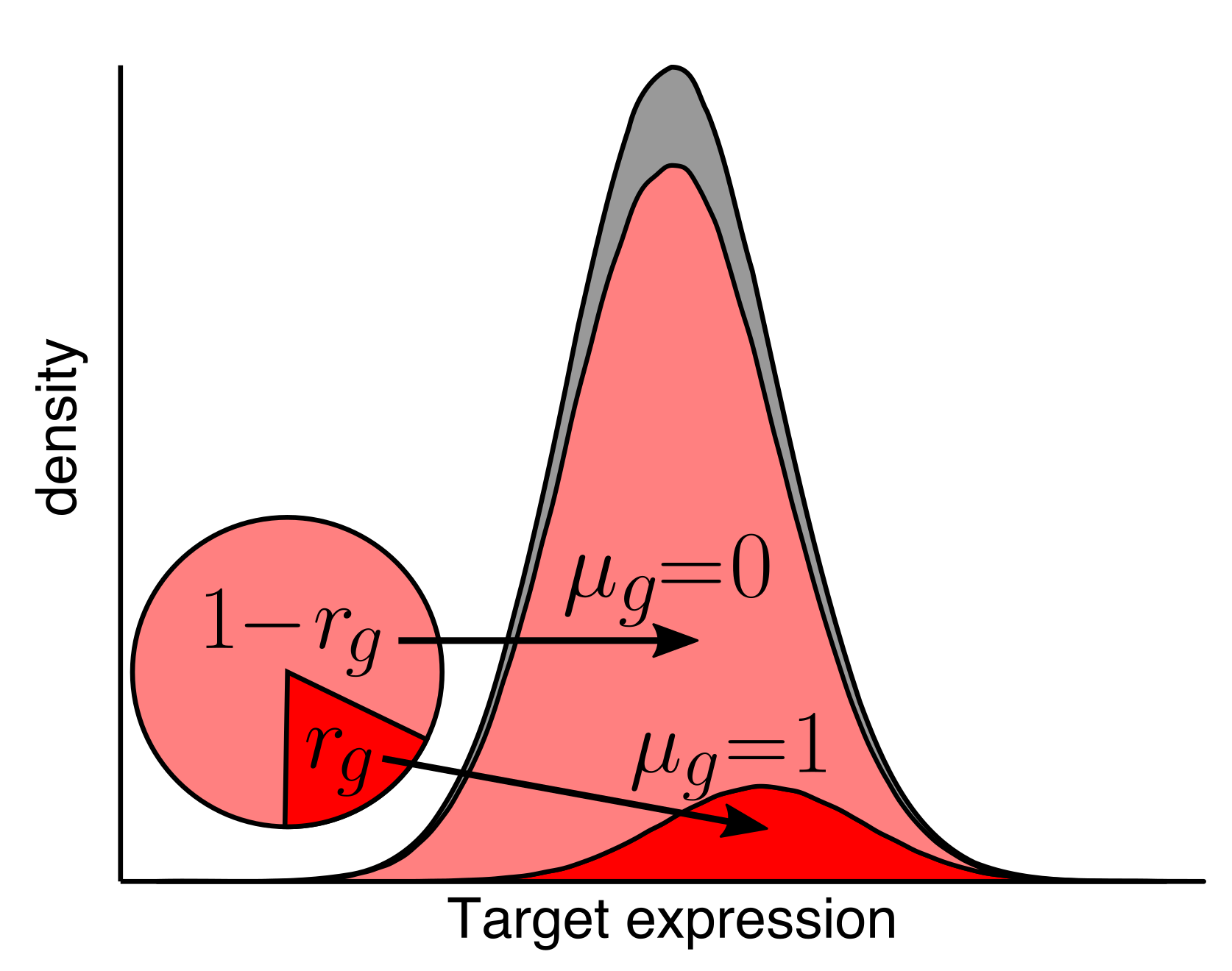
Development of genomics tools
As we and others develop new genomics technologies, new computational approaches are needed to make the most of the data. We create algorithms and tools to process, analyze, and understand genomics data.
Funding
We are grateful to the funders who support our research.





